
This guide contains general technical information for viral growth, propagation, preservation, and application.
Getting Started With an ATCC Viral Strain
Viral Replication and Propagation
Growth Media for Tissue Culture-adapted Viruses
Preservation
Biosafety and Disposal
Viral Authentication and Viability Testing
Viral Applications
Appendix
ATCC Reagents for Virus Expansion
Glossary
References
Get a copy of the ATCC Virology Culture Guide to help with your research
Download the GuideGetting started with an ATCC viral strain
ATCC viral strains are predominantly shipped either frozen on dry ice in plastic cryopreservation vials, or as lyophilized materials within glass ampoules or serum vials. Upon receipt of frozen material, either thaw and transfer the viral agent to an appropriate propagation host, or briefly store the frozen material between -70°C and -80°C to allow time to seed host cells. However, please note that the viability of some materials may decline at temperatures above -120°C. Upon receipt of freeze-dried strains, reconstitute cultures with sterile, double-distilled water and add the rehydrated material to an appropriate propagation host. If this is not possible, store the vials in liquid nitrogen vapor phase (below -120°C).
Product sheet
A product sheet that contains detailed information on the production host and recommendations for infection are provided for each ATCC virology strain. This product sheet, as well as additional information, can be found on the ATCC website or can be requested from the ATCC Technical Service Department.
Viral taxonomy
Influenza ultra-structure courtesy of Jordan Douglas, CDC
Viruses are placed into taxonomic groups based on characteristics including morphology, genome type, and host organism. Viral agents can significantly vary in size, often ranging between 20 and 300 nanometers in diameter. They also vary in structure, including helical, icosahedral, prolate, enveloped, and complex morphologies.
In addition to unique morphological structures, viruses also vary in genomic structure. Unlike other microorganisms that have double stranded DNA as genomic material, viral genomes can be composed of double-stranded DNA, single-stranded DNA, double-stranded RNA, or single-stranded RNA. Single-stranded RNA viruses can be further described as positive sense, negative sense, or ambisense. For more detailed information on the various morphological and genomic types, please refer to the glossary.
Changes in taxonomy or further analysis of viral strains may lead to a change in nomenclature. Taxonomic nomenclature as well as the common name can be found on the product sheet. Further information on viral nomenclature is available from the International Committee on Taxonomic Viruses.
Viral replication
Viruses are pathogenic intracellular organisms requiring living cells in order to multiply. The virus life-cycle can be divided into three major steps: attachment, assembly, and release. Generally, infection is established when the virus binds to a specific cellular receptor and enters the host cell. Upon cellular entry, host proteins are recruited to assist with viral replication. Once viral structural proteins are generated, new viruses assemble within the cell. Depending on the nature of the viral agent, the replication and assembly process can vary in cellular location and process. Following viral assembly, new infectious particles either remain cell-associated or exit the cell via lysis or virus shedding.
Preparation of propagation host and reagents
In advance, prepare the propagation host and associated reagents necessary for viral propagation. Information for the preparation of these products is available on the provided product sheet.
Opening glass ampoules containing frozen material
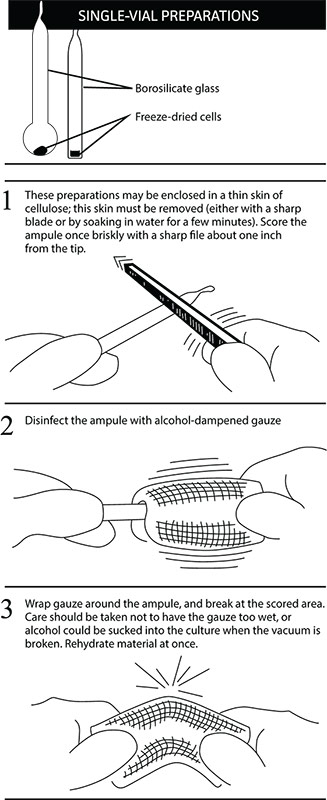
Overview
All cultures should be considered potentially hazardous and should be opened by individuals trained in microbiological techniques. Work should only be carried out in facilities with containment requirements appropriate for the biosafety level of the cultures. The handling or opening of glass ampoules containing virus material must be performed in a biological safety cabinet. Ensure that all empty vials are sterilized before disposal.
- Disinfect the outside of the ampoule with freshly prepared 70% ethanol or dip it into a beaker of freshly prepared 70% ethanol.
- To recover the material from the glass ampoule, score the neck of the ampoule with a sterile, small file.
- Wrap the ampoule within several folds of a sterile towel or gauze to dry residual ethanol.
- Working in a biological safety cabinet, hold the vial upright and snap open the vial. Ensure that your gauze does not become too wet with ethanol, or ethanol could be sucked into the culture when the vacuum is broken. Propagate the virus immediately.
Initiating frozen cultures
Tissue culture-adapted strains
- In advance, prepare the cell growth medium for growing the host cell line. Additionally, prepare the virus growth medium for virus propagation as noted on the product sheet. Viral growth medium is usually supplemented with a lower percentage of serum than cell growth medium, often ranging between 2-10% depending on the virus (See NOTE 1). Ensure that both the cell and viral growth media are equilibrated for temperature and pH.
- Initiate the recommended production host, and grow the monolayer to an appropriate confluency.
- Prior to thawing the frozen virus stock, check the virus titer listed on the lot specific certificate of analysis to determine the amount of virus needed. Thaw the vial of frozen virus via gentle agitation in a water bath set at 37°C. Thawing will be rapid (approximately 2 minutes or until all ice crystals have melted).
- Remove the vial from the water bath and decontaminate the outer surface using 70% ethanol. Follow strict aseptic conditions in a biological safety cabinet for all further manipulations.
- Prepare the viral stock in base medium (without serum) unless specified.
- Remove the cell growth medium from the cell culture, wash the monolayer of cells once or twice with phosphate buffered saline (PBS), and inoculate with the prepared viral stock to provide an optimal multiplicity of infection (MOI) as indicated on the product sheet, if applicable.
- Propagate the virus according to the recommended infection conditions as described on the product sheet. For additional information, refer to the chapter Viral Replication and Propagation.
NOTE 1:
Viral growth medium prepared for tissue culture-adapted strains of Influenza should not be supplemented with Fetal Bovine Serum (FBS).
Strains propagated in chicken eggs
- In advance, prepare chicken eggs by incubating eggs under the recommended temperature and atmospheric conditions. Additionally, all eggs should be candled to ensure viability and properly developed embryos. Healthy embryos will have an air sac, well-developed blood vessels, and observable movement.
- Prior to thawing the frozen viral preparation, check the certificate of analysis for lot specific titer information. Thaw the frozen virus vial via gentle agitation in a water bath set at 37°C. Thawing will be rapid (approximately 2 minutes or until all ice crystals have melted).
- Remove the vial from the water bath and decontaminate the outer surface using 70% ethanol. Follow strict aseptic conditions in a biological safety cabinet for all further manipulations.
- Inoculate eggs using the inoculation procedure described in the chapter Viral Replication and Propagation.
Initiating lyophilized cultures
- Open vial according to enclosed instructions. Instructions are also available within the instructional guide, "Reviving Freeze-Dried Microorganisms."
- Aseptically add an appropriate volume of sterile double-distilled water at room temperature. Mix well.
- Dilute the sample in base medium (without serum) unless specified, and inoculate host cells to provide an optimal MOI as indicated on the product sheet, if applicable.
Viral replication and propagation
Propagation host range
For tissue culture-adapted strains, the appropriate selection and processing of cell cultures is important for successful viral isolation, titer, and infectivity.2 Typically, viruses can only infect a limited number of hosts, known as the host range. This is best explained by a “lock and key” mechanism as certain proteins on the viral surface must fit specific receptor sites on the host cell surface. When using ATCC viral strains, the recommended host is indicated on the provided product sheet. For customer convenience, ATCC Cell Biology holdings include cell lines for the propagation of viruses (Table 1, Appendix).
Table 1: Examples of propagation hosts for tissue culture-adapted viruses
| ATCC number | Product name | ATCC number | Propagation host |
|---|---|---|---|
| 53592 | Chlamydophila pneumonia* | CCL-23 | HEp-2 |
| VR-129B | Encephalomyocarditis | CCL-81 | Vero |
| VR-1492 | Human herpesvirus 4 (Epstein-Barr virus) | N/A | B95-8 |
| VR-260 | Humans herpesvirus 1 | CCL-81 | Vero |
| VR-734 | Human herpesvirus 2 | CCL-81 | Vero |
| VR-1367 | Human herpes 3 | CCL-171 | MRC-5 |
| VR-538 | Human herpesvirus 5 | CCL-171 | MRC-5 |
| VR-93 | Human parainfluenza | CCL-7.1 | LLC-MK2 Derivative |
| VR-26 | Human respiratory syncytial virus | CCL-23 | HEp-2 |
| VR-1540 | Human respiratory syncytial virus | CCL-23 | HEp-2 |
| VR-1580 | Human respiratory syncytial virus | CCL-23 | HEp-2 |
| VR-283 | Human rhinovirus 16 | CRL-1958 | H1-HeLa |
| VR-95 | Influenza A virus (H1N1) | N/A | SPF CE |
| VR-897 | Influenza A virus (H1N1) | N/A | SPF CE |
| VR-1469 | Influenza A virus (H1N1) | CCL-34 | MDCK |
| VR-1520 | Influenza A (H1N1) | CCL-34 | MDCK |
| VR-544 | Influenza A virus (H3N2) | N/A | SPF CE |
| VR-1679 | Influenza A virus (H3N2) | CCL-34 | MDCK |
| VR-1535 | Influenza B virus | CCL-34 | MDCK |
| VR-1583 | JC polyomavirus | CRL-1651 | COS-7 |
*For historical reasons and because of similar requirements for handling, Chlamydia and Rickettsia are included in this guide
Viral propagation
Propagation in cell culture
A number of ATCC viruses are propagated in cell culture. Typically, propagation hosts are grown in tissue culture vessels (such as T flasks) using media and reagents specified for the host cell line. Most cell lines are seeded the day prior to setting up an infection and should not be seeded more than two days in advance, nor passaged more than 9 times prior to infection. In addition to setting up cells for infection, negative control cells should also be set up to monitor cellular health.
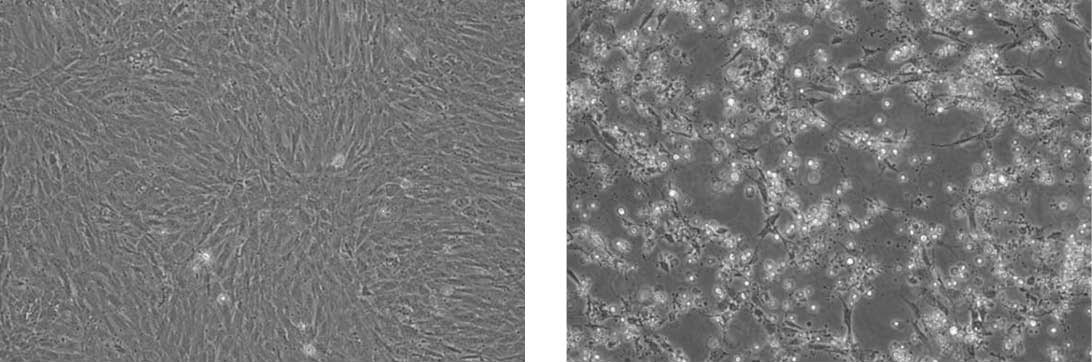
Propagation of Feline infectious peritonitis virus (ATCC VR-2004) in CRFK cells (ATCC CCL-94). The panel on the left are uninfected CRFK cells. The panel on the right are infected CRFK cells exhibiting CPE.
- Identify the recommended propagation host as indicated on the product sheet. Plan to use the propagation host.
- Prepare the cell growth medium for growing the host cell line.
- Prepare the virus growth medium as recommended on the product sheet.
- One to two days prior to inoculation seed the host cells. Be sure to include a vessel that will not be inoculated with virus to serve as a negative control.
- Allow cells to reach the appropriate confluency. (See NOTE 2)
- Prior to thawing the frozen virus stock check the virus titer listed on the certificate of analysis. Quickly thaw the virus in a 37°C water bath.
- Dilute the virus stock in the appropriate volume of viral growth medium.
- Remove the cell growth medium from the cell culture flasks.
- Inoculate the diluted virus to provide an optimal MOI as indicated on the product sheet.
- Incubate tissue cultures under the appropriate temperature and atmospheric conditions for the recommended incubation period.
- After the recommended incubation time period, check the flask for cytopathic effects (CPE) when applicable.
NOTE 2:
The required confluency of the host cell line will differ between viruses.
Propagation in chicken eggs
Several ATCC virus holdings, such as influenza, are propagated in embryonated chicken eggs. There are many advantages to culturing in eggs, including easy care, quick viral replication, and an inherently aseptic environment. When culturing in embryonated eggs, ensure that eggs are viable, have properly developed blood vessels and air sacs and are obtained from pathogen-free flocks. Depending on the virus, different inoculation routes and/or organs may be used to cultivate the agent. These include the allantoic cavity, the amniotic cavity, the chorioallantoic membrane (CAM), and the yolk sac (Table 2). Below, we describe the procedures for allantoic cavity, amniotic cavity, and CAM inoculation. For the following procedures, ATCC recommends performing viral inoculation and harvest within a biological safety cabinet. For additional information on the inoculation or propagation of embryonated eggs, contact ATCC technical services.
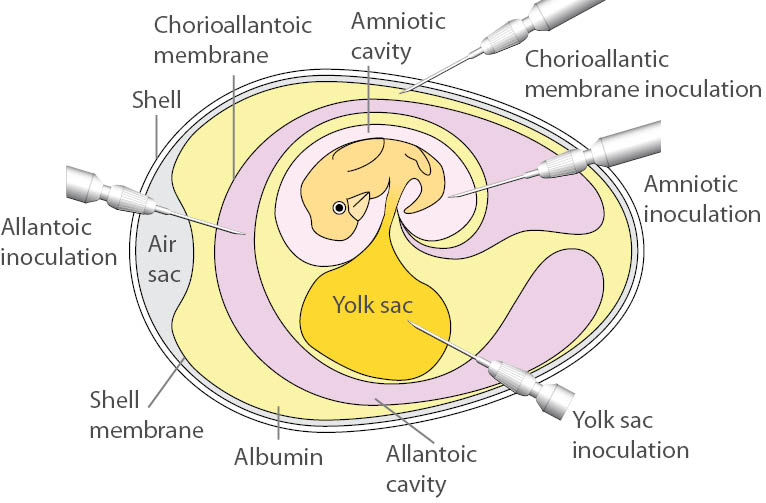
- Allantoic cavity inoculation
- For allantoic cavity inoculation, most viruses require 10-day old embryos. While candling the eggs, draw a pencil line around the air sac. Using an 18.5 gauge needle, poke a hole into the air sac.
- Using a 1.0 mL syringe fitted with a 22.5 gauge needle, inject the viral inoculum through the hole, into the allantoic cavity, being careful not to stick the embryo.
- Seal the hole with tape or wax.
- Incubate the inoculated eggs under conditions recommended for viral replication. Candle eggs 12-18 hours after inoculation and discard eggs that are non-viable.
- To harvest allantoic fluid, refrigerate eggs for at least 2 hours post-incubation. Once the embryo is no longer viable, open the egg by tapping on the shell just above the air sac until the shell breaks.
- Use sterile scissors to cut away the shell around the air sac.
- Aspirate the allantoic fluid with a syringe or a pipette. Usually 5 mL can be harvested from each egg; this is considered infectious material.
- Amniotic cavity inoculation
- While candling the eggs, make a small hole in the side of the egg. Create the hole slowly as it is easy to accidently stick the embryo.
- Using a 1.0 mL syringe, insert the needle and syringe containing the viral inoculum into the egg until the amniotic sac moves slightly. The needle is then thrust through the membrane and the fluid is injected slowly.
- Immediately seal the hole in the shell with tape or wax.
- Incubate the inoculated eggs under conditions recommended for viral replication.
- Candle eggs 12-18 hours after inoculation and discard eggs that are non-viable.
- To harvest amniotic fluid, refrigerate eggs for at least 2 hours post-incubation. Once the embryo is no longer viable, open the egg by tapping on the shell just above the air sac until the shell breaks.
- A needle and syringe must be used to aspirate the fluid. The needle may be inserted directly into the amniotic sac, or the embryo may be carefully removed from the egg and placed in a petri dish. Typically, one embryo will yield about 1.0 mL of fluid.
- Chorioallantoic membrane (CAM) inoculation
- Mark an area free from large blood vessels on the side where the embryo is located and the area over the air sac.
- Make one hole on the side and one over the air sac. Puncture the shell taking care not to damage the CAM.
- Gently apply suction to the hole over the air sac. Do this over an egg candler to see the CAM drop and the new air sac form.
- Add 1.0 mL of the viral inoculum on the dropped membrane and rotate the egg to distribute the inoculum over the membrane.
- Seal the hole with nail polish.
- Candle inoculated eggs daily. Any embryos that die within 24 hours should be discarded.
- To harvest, refrigerate eggs for at least 2 hours. Once the embryo is no longer viable, make a tape handle over the area of inoculation. To do so, cut a piece of tape, without joining either end, join the middle of the piece together leaving both ends free to adhere to the egg shell with a middle piece that is no longer sticky, forming a handle sticking up from the egg shell.
- Cut off the top half of the eggshell, including the infected area, and gently remove the CAM, which is attached to the shell.
- Place the infected CAM in a tissue culture dish with PBS, spread the membrane flat against the dish, and place the dish on a dark surface to facilitate counting of pocks.
Table 2: Examples of ATCC viruses propagated in embryonated chicken eggs
| Product name | Egg age (days) | Inoculation route | Incubation (days) | Optimal temperature (°C) | Death of embryo |
|---|---|---|---|---|---|
| Influenza A virus | 10-11 | Allantoic | 2-3 | 33-35 | - |
| Influenza B virus | 10-11 | Allantoic | 2-3 | 33-35 | - |
| Sendai virus | 10-11 | Allantoic | 2-3 | 35-37 | + |
| Rabies virus | 7 | Yolk | 9-10 | 36.5 | - |
Propagation of common viruses and Chlamydia
Human herpesvirus 4 (Epstein-Barr virus)
Epstein-Barr virus (EBV) is a universal human pathogen commonly transmitted via saliva.3 Primary infections among adolescents and young adults result in mononucleosis, a condition characterized by fever and the swelling of lymph tissues. Following infection, EBV remains in a latent stage in most adults. In rare cases, the virus may reactivate and contribute to neoplastic disorders such as Burkitt’s lymphoma or post-transplant lymphoproliferative disorder (PTLD).4 To propagate EBV, the virus is often harvested from tumors growing on patients with Burkitt’s lymphoma, or obtained from a biological resource center such as ATCC, and further grown in human lymphoblastoid cells.
ATCC number: VR-1492, VR-603, VR-602
Recommended host: Human lymphoblastoid cells
Preparation: Infected tissue culture with DMSO and FBS
Incubation: 5-15 days at 37°C
Atmosphere: 5% CO2 in an air atmosphere
Effect: Polykaryocyte formation and cell swelling
Influenza
Image of Influenza courtesy of Dr. Erskine L Palmer and ML Martini, CDC
Influenza viruses are highly contagious, enveloped, airborne pathogens that cause acute febrile illness, fatigue, and respiratory infection. There are three types of influenza including influenza A, B, and C, which are distinguished by the presence of specific, core nucleoproteins. Of these viral types, influenza A is considered the most pathogenic. To propagate influenza, the virus is often cultured in the allantoic fluid of embryonated chicken eggs. However, some strains are propagated in amniotic fluids or have been adapted for growth in tissue culture.
ATCC number: VR-1469, VR-95, VR-1520, VR-544, VR-897, VR-1679, VR-1807, VR-1811, VR-1813
Recommended host: Pathogen-free embryonated chicken egg, 10-11 days old, or MDCK cells (ATCC CCL-34)
Preparation: Infected chicken egg allantoic fluid, infected tissue culture
Incubation: 2-3 days at 34°C for influenza propagated in chicken eggs (CE), 33-35°C for influenza propagated in tissue culture (TC). Harvest when CPE has progressed throughout the monolayer.
Atmosphere: An air atmosphere for CE, 5% CO2 atmosphere for TC. CO2 level is determined by media formulation.
Effect: Hemagglutination of chicken red blood cells, cell rounding sloughing in tissue culture.
Respiratory syncytial virus
Image of Respiratory Syncytial Virus courtesy of Dr. Craig Lyerla, CDC
Respiratory syncytial virus (RSV) is a major cause of respiratory illness in young children, resulting in pneumonia and bronchiolitis. In adults, RSV often manifests symptoms similar to the common cold. This virus is commonly transmitted when droplets containing the virus are aerosolized by coughing or sneezing.
ATCC number: VR-1540, VR-1540P, VR-26, VR-1580, VR-1803
Recommended host: HEp-2 (ATCC CCL-23) and HeLa (ATCC CCL-2) cells
Preparation: Infected tissue culture fluid and cell lysate
Incubation: 5-12 days at 37°C
Atmospherte: 5% CO2 in an air atmosphere
Effect: Syncytia formation
Chlamydia
Species within the Chlamydia genus are obligate intracellular bacterial pathogens commonly transmitted through sexual activity. Common symptoms associated with Chlamydia species include conjunctivitis, pelvic inflammatory disease, and infection of the reproductive organs. These bacterial species are commonly categorized with viruses as they have similar propagation properties. Often it is recommended that the Chlamydia stock be sonicated first before inoculating cells, and infection may be enhanced by centrifugation after inoculation. Follow the specific instructions on the product sheet for preparing the cells.
ATCC number: VR-123, VR-346, VR-347, VR-573, VR-879, VR-885, VR-886, VR-887
Recommended host: McCoy cells (ATCC CRL-1696)
Preparation: Infected tissue culture fluid and cell lysate
Incubation: 2-3 days at 35-37°C
Atmosphere: 5% CO2 in an air atmosphere
Effect: Intracellular inclusion bodies visualized by fluorescent staining with genus or species specific conjugated monoclonal antibodies.
Viral titering
Plaque assay
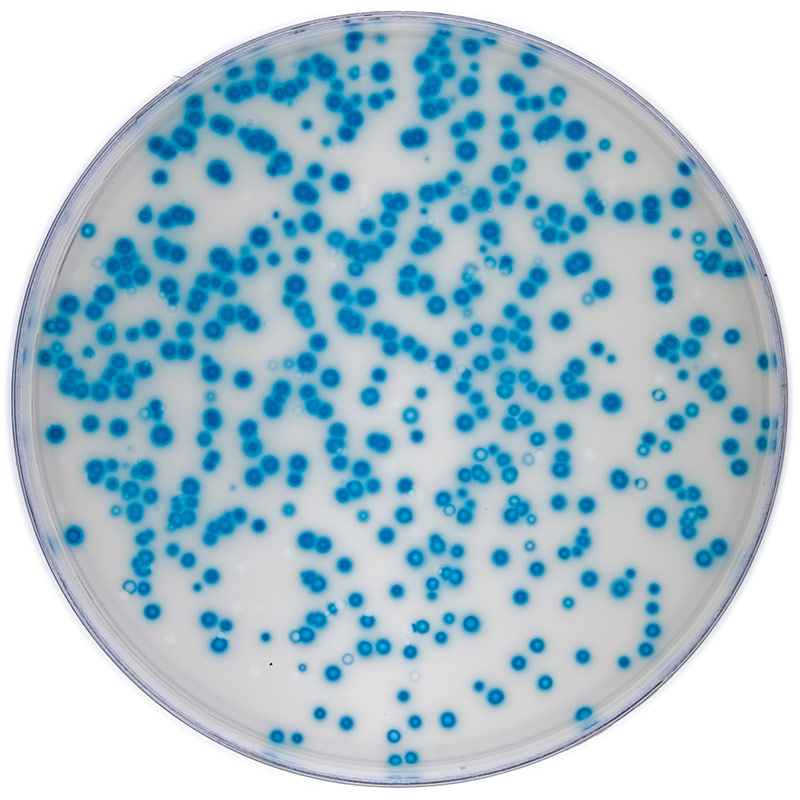
Plaque assay of a tissue culture-adapted viral strain.
Calculating viral titer is necessary in order to determine viral infectivity. This can be determined by a plaque assay or through calculating the infectious dose. The plaque assay was initially developed to count and measure the infectivity level of bacteriophages. This technique has since been modified for use in animal virology, and has been a reliable determination of titer for a number of tissue culture-adapted viruses. The basis of this assay is to measure the ability of a single infectious virus to form a plaque on a cell culture monolayer. A plaque is developed as part of the viral infection cycle, where following viral replication, the host cell dies.5
- Grow the host cells in wells with the recommended growth medium for the cell line. Allow the cells to reach the appropriate confluency.
- Remove the growth medium and wash with Dulbecco's Phosphate-Buffered Saline (DPBS). Add diluted virus to each well, using multiple wells per dilution.
- Incubate dishes for 1-2 hours to allow for viral adsorption.
- Remove the inoculum and wash with basal medium, if applicable.
- Overlay the cells with overlay medium. Incubate for a length of time appropriate for infection. Remove overlay, if applicable.
- Observe the cell monolayers daily for the presence of foci or plaques.
- Count the number of plaques on plates with 20 or more plaques. The average amount of plaques per Petri dish multiplied by the virus dilution gives the number of plaque forming units (PFU) per volume of inoculum. This number may be expressed as the titer of the virus. (See NOTE 3)
NOTE 3:
This is a general procedure used to determine the potency of a virus. Please refer to the following references for more details on this procedure: A Manual of Basic Virological Techniques, published by Prentice-Hall Inc.,1 or Virology Methods Manual, published by Academic Press. (1996).5
Tissue culture infectious dose (TCID)
Viral titer can be determined in vitro by calculating the infectious dose. For tissue culture-adapted strains, this calculation is ascertained through an endpoint dilution assay in cell culture. The most reproducible endpoint of the dilution assay is the dilution of the virus that will produce a pathological change in 50% of the cell cultures inoculated. This number is expressed as 50% the infectious dose, or TCID50, which is analogous to the calculation for lethal dose 50. The accuracy of this method is related to the number of replicates at each dilution.5
- Prepare a cell culture plate with the recommended cell line for viral propagation (preferably the same cells that the virus being tested was grown in).
- Incubate the plate under the appropriate conditions for cell growth until cells reach an optimum density for infection.
- Prepare viral dilutions in base medium.
- Remove cell growth medium from plate, the monolayer may be washed with remove any inhibitory agents.
- Inoculate at least 3 wells with each dilution. Be sure to inoculate wells with base medium to serve as negative controls. Use a fresh, sterile pipette for each dilution.
- Allow the plate to incubate for 1 to 2 hours under conditions suitable for virus adsorption.
- Add viral growth medium and incubate the plate under conditions suitable for the virus and observe all wells daily. Record results for each well daily.
- The endpoint is determined when the CPE or immunofluorescence assay (IFA) read-out appear the same per dilution for 3 separate readings.
- The titer is calculated using the method of Reed and Muench.6 A titer expressed as 10(3.0)TCID50/0.2 mL in 3 days in XXXX cell line may be translated as: 0.2 mL of virus diluted at 1:1000 will infect 50% of the cells in 3 days when using XXXX cell line. (See NOTE 3)
The TCID50 can be converted to plaque forming units (PFU) using the Poisson distribution. This conversion is an estimate based on the rationale that the limiting dilution, which would infect 50% of the cell layers challenged, would be expected to produce a single plaque in a cell monolayer. However, ATCC recommends that the actual number of PFUs be determined empirically.
To estimate PFU from TCID50, the Poisson distribution can be applied; P(o) is the proportion of negative tubes and 'm' is the mean number of infectious units per volume (PFU/mL), P(o)=e(-m). For any titer expressed as TCID50, P(o)=0.5. Thus, e(-m)=0.5 and m= -ln 0.5, which is ≈0.7. Therefore, one could multiply the TCID50 titer (per mL) by 0.7 to predict the mean number of PFU/mL. For example, one can assume that material with a TCID50 of 1 × 105 TCID50/mL will produce approximately 0.7 × 105 PFU/mL. When applying this calculation, remember that the estimated mean will only be valid if the changes in the protocol required to visualize plaques do not alter viral expression as compared to conditions used to determine TCID50.
Chicken embryo infectious dose (CEID)
For viruses normally propagated in chicken eggs, such as influenza virus, viral titer is calculated as the chicken embryo infectious dose (CEID). Viral cultures are serially diluted and are used to inoculate embryonated chicken eggs. Following incubation, allantoic fluid is harvested from each egg at all dilutions and virus titer is determined by the appearance of hemagglutination. A positive hemagglutination reaction indicates the virus is present at that dilution.
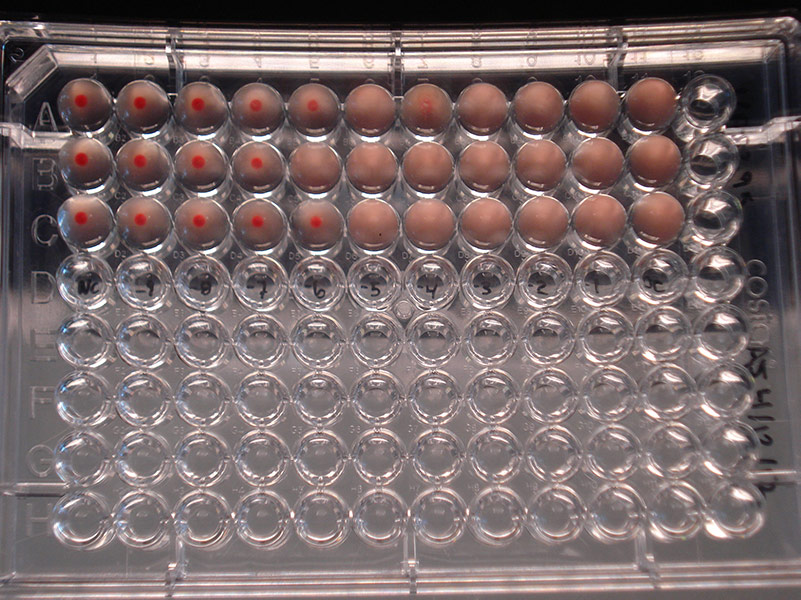
Hemagglutination assay depicting Influenza B virus (ATCC VR-295) at dilutions varying from 10-9 to 10-1. Left column = Negative control; Columns 2-10 = Dilutions 10-9 to 10-1; Column 11 = Positive control
- Order embryonated chicken eggs of the proper age, 8-12 days old depending on the virus. When the eggs arrive, place them in an incubator at an appropriate temperature with a dish of water close by to provide moisture to prevent the eggs from drying out.
- When you are ready to inoculate, candle the eggs to locate the air sac and embryo. Using a pencil, outline the air sac area and indicate where the embryo’s eye is located. In a biological safety cabinet, punch a hole into the shell within the defined air sac area, on the opposite side of the egg from the embryo’s eye. Inoculate the allantoic fluid of each egg with the serial dilution of virus. Seal the hole with nail polish and incubate the eggs near a pan of water at the proper temperature as recommended on the provided product sheet. (See NOTE 4)
- Candle the eggs 24 hours post-inoculation to check for viral-induced death. Place any of the dead eggs into a biohazard bag, seal the bag with autoclave indicator tape and refrigerate to await autoclaving. Return the rest of the eggs to the incubator.
- Following the recommended incubation period, harvest the virus. Depending upon the virus, death of the embryo may have occurred. By this time, a viable embryo will have grown much larger, will be more active, and the network of blood vessels will be more visible. A dead embryo will not move and the egg may or may not be as transparent and the blood vessels will have mostly disappeared. For viable embryos, sacrifice the embryo prior to viral harvesting by placing the egg at 4°C for at least 2 hours.
- Prepare a 0.5% suspension of chicken red blood cells using phosphate buffered saline (PBS) while the infected eggs are chilling in a biological safety cabinet. Use sterile forceps to break the top of the shell open and pull back the allantoic membrane. Place all egg fragments, including parts of the shell into a double biohazardous materials bag. Harvest the allantoic fluid from each egg by using a pipette to collect the fluid into a different well of a rounded-bottom 96-well plate for each egg, being sure to use a new pipette for each dilution.
- Add a 1:1 ratio of the 0.5% red blood cell suspension to allantoic fluid in each tube. Mix by gently tapping and allow the red blood cells to settle to the bottom of the wells. After 30-45 minutes at room temperature, read the hemagglutination assay by recording the presence of a button of cells or the presence of a lattice formation at the bottom of the tube. A negative result is seen by the button of red blood cells at the bottom of the rounded well. A positive result is seen by the formation of the lattice formation at the bottom of the rounded well. Once the results are properly recorded, place the plate in a biohazard bag to await proper decontamination.
- The end point is taken to be the highest dilution of virus suspension that produces a positive result. HA = 1:640 means that the virus was titered by a hemagglutination assay (HA) and the endpoint for the assay was a dilution of 1:640. (See NOTE 3)
NOTE 4:
A viable egg has a series of small blood vessels within the allantoic membrane, which can be seen during candling. Additionally, the embryo’s eye will look like a black spot, which will move.

Growth media for tissue culture-adapted viruses
Media for culturing propagation hosts
For the propagation of tissue culture-adapted viruses, it is important to grow and maintain the host cell line in an appropriate medium. The recommended media for all host cells is indicated on the product sheet for the host cell line and can also be found online at www.atcc.org.
Cell culture media are complex mixtures of salts, carbohydrates, vitamins, amino acids, metabolic precursors, growth factors, hormones, and trace elements. The requirements for these components vary among cell lines. Carbohydrates are supplied primarily in the form of glucose. In some instances, glucose is replaced with galactose to decrease lactic acid build-up, as galactose is metabolized at a slower rate. Other carbon sources include amino acids (particularly L-glutamine) and pyruvate.
In addition to nutrients, the medium helps maintain the pH and osmolality in a culture system. The pH is maintained by one or more buffering systems; CO2/sodium bicarbonate, phosphate, and HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) are the most common. Sera will also buffer a complete medium. Phenol red, a pH indicator, is added to the medium to colorimetrically monitor changes in pH. Media commonly used in the propagation of tissue culture-adapted strains include the following:
Eagle’s Minimum Essential Medium (EMEM) ATCC’s modification of EMEM (ATCC 30-2003) contains Earle’s balanced salt solution, non-essential amino acids, L-glutamine, and sodium pyruvate. It is formulated with a reduced sodium bicarbonate concentration (1,500 mg/L) for use with 5% CO2. Because EMEM is a simple medium, it is often fortified with additional supplements or higher levels of serum.
Dulbecco’s Modified Eagle’s Medium (DMEM) has roughly twice the concentration of amino acids and four times the amount of vitamins as EMEM, as well as ferric nitrate, sodium pyruvate, and some supplementary amino acids (though not all nonessential amino acids). The original formulation contained 1,000 mg/L of glucose, but in the more commonly used variations this amount was increased to 4,500 mg/L. ATCC DMEM (ATCC 30-2002) has 4,500 mg/L of glucose and a reduced sodium bicarbonate concentration (1,500 mg/L) for use with 5% CO2.
RPMI-1640 ATCC’s RPMI-1640 (ATCC 30-2001) was modified to contain higher amounts of glucose (4,500 mg/L), sodium pyruvate, and HEPES buffer. It also contains a reduced concentration of sodium bicarbonate (1,500 mg/L) for use with 5% CO2.
Leibovitz’s L-15 Medium (ATCC 30-2008) is formulated for free gas exchange with atmospheric air. The standard sodium bicarbonate/CO2 buffering system is replaced by a combination of phosphate buffers, free-base amino acids, higher levels of sodium pyruvate, and galactose. A CO2 and air mixture is detrimental to cells when using this medium for cultivation. However, cell cultures can be grown in CO2 incubators with L-15 medium provided there is no exchange between the air in the culture vessel with that of the incubator (ie, caps of flasks are tightly closed).
Media formulations
The formulations for media used by ATCC can be found online. Please note that there are cell lines in the collection used for viral propagation that may require media that is not currently sold by ATCC.
Media supplements
The growth media recommended for host propagation may require the addition of components not already available in the base medium. These components may include hormones, growth factors, or serum.
After supplements have been added to the base medium, the shelf life of the medium should be determined on a case-by-case basis. Media containing serum, antibiotics, and/or antimycotics tend to degrade faster than base media alone. Media containing supplements should not be frozen as this may cause certain compounds to precipitate out of solution; media should be stored at 2°C to 8°C. For additional information regarding the preparation, storage, or usage of specific additives, contact your local supplier or consult with the manufacturer’s product information sheet.
Serum
Serum can serve as a source of growth factors, proteins, vitamins, hormones, carbohydrates, lipids, amino acids, minerals, and trace elements. The exact composition of serum is unknown and varies from lot to lot, although lot- to-lot consistency has improved in recent years.
Sera from fetal bovine sources are commonly used to maintain cell cultures in preparation for viral infection. Fetal serum is a rich source of growth factors and is appropriate for the growth of fastidious cells. It is often supplied at a concentration of 2-10%. (See NOTE 5)
Unfortunately, naturally derived products from animals, such as sera, may contain adventitious microorganisms. All reputable suppliers routinely test their products for infectious viruses by several methods including fluorescent antibody labeling, cytopathic effect, and hemadsorption. These products are also screened for the standard microbial contaminants such as bacteria, fungi, and mycoplasma. To reduce the risk of any possible contamination, ATCC recommends that all serum should be triple filtered through 0.1 µm sterile filters before use.
NOTE 5:
For viral inoculation, ATCC recommends decreasing the percentage of serum to 2% as it can interfere with viral attachment. This can vary from strain to strain, and is not applicable for influenza viruses.
ATCC offers the following types of animal sera:
- Fetal Bovine Serum (also known as fetal calf) – ATCC 30-2020
- Fetal Bovine Serum qualified for embryonic stem cells – ATCC SCRR-30-2020
- Iron-supplemented Calf Bovine Serum – ATCC 30-2030
These products are rigorously tested for adventitious infective agents and sourced only from U.S. herds. Further, each lot is tested for its ability to support cell culture growth and is the same sera used in ATCC labs.
- Storage
Do not store serum at temperatures above -20°C for any length of time. Avoid any repeated freeze-thaws by dispensing and storing sera in aliquots. Additionally, ensure that sera are stored away from direct light. - Thawing
The following procedure is used to thaw serum:- Place the frozen serum in a refrigerator at 2°C to 8°C overnight.
- Put the bottles in a 37°C water bath and gently agitate occasionally to mix the solutes that tend to concentrate at the bottom of the bottle.
- Turbidity and precipitates
All sera may retain some fibrinogen. Because external factors may initiate the conversion of fibrinogen to fibrin, flocculent material or turbidity may be observed after the serum is thawed. The presence of this material does not alter the serum’s performance. If the presence of flocculent material or turbidity is a concern, it can be removed by filtration through a 0.45 µm filter.
A precipitate can form in serum when incubated at 37°C or higher for prolonged periods. This is often mistaken for microbial contamination. This precipitate may include crystals of calcium phosphate, but this does not alter the performance of the serum as a supplement. Heat inactivation of sera can also cause the formation of precipitates.
Additives and cryoprotectants
To preserve viral strains by cryopreservation, ATCC may use a mixture of FBS and DMSO (ATCC 4-X) or glycerol. Some viruses require a specialized mixture of cryoprotectants or additives. For more information, refer to the cryopreservation section of the chapter entitled Preservation.

Preservation
There is no single procedure that is optimal for the preservation of all animal viruses. Both the type of agent and recommended growth conditions of the culture influence the method of cryopreservation, and the determination of these ideal conditions may require some experimentation. There are many advantages of preservation that far outweigh the required investment in equipment and reagents for continuous in vitro expansion. These advantages include:
- Overall safety of viral stocks against loss due to equipment failure or contamination by other microbial organisms.
- Elimination of time, energy, and material costs associated with the maintenance of viral strains not currently in use.
- Insurance against phenotypic drift due to genetic instability and/or selective pressures.
- Creating a standard reagent that can be used for a series of experiments.
The methodologies used at ATCC have proven successful for a large number of organisms. Currently, ATCC only uses cryopreservation methods to preserve viral strains. However, several viral strains in the collection are available as freeze-dried (lyophilized) cultures. The basic methodology for the cryopreservation and lyophilization of viruses are described in the subsequent sections. For historical reasons and because of similar requirements for handling and preservation, Chlamydia and Rickettsia species are also included here.
Cryopreservation
Overview
Freezing a viral suspension often results in a decrease in viability and titer.11,12 For cell-associated pathogens, such as Chlamydia and Rickettsia, as the suspension is cooled below the freezing point, ice crystals begin to form and the concentration of solutes in the suspension increases resulting in damage of host cell structures. This can be minimized if water within the cells is allowed to escape by osmosis during the cooling process; a slow cooling rate, generally -1°C to -10°C per minute, facilitates this progression. However, as host cells lose water, they shrink in size and will quickly lose viability if they surpass a minimum threshold volume. The addition of cryoprotectant agents, such as glycerol or dimethylsulfoxide (DMSO), will mitigate these effects.13,14
The viability of viruses that are not cell-associated is best maintained by rapid freezing. In this method, samples are quickly frozen in a dry ice slurry and stored in liquid nitrogen vapor or within a mechanical freezer at -80°C.
Overall, there are numerous factors that can affect the viability of recovered viruses. These critical parameters can include the composition of the cryoprotectant and the viral titer. For cell-associated viruses, obtain optimal cell viability upon recovery by modifying the cryopreservation procedure for each viral strain. Contact ATCC for more information on the cryopreservation of viral strains.
Freeze medium
Glycerol and DMSO are the most common cryoprotectant agents. To preserve viral strains, ATCC commonly uses a mixture of FBS and DMSO or glycerol. When employing these cryoprotectants, use only reagent-grade DMSO or glycerol. Store both in aliquots protected from light. ATCC offers DMSO (ATCC 4-X) that has been thoroughly tested for use.
Some viruses require specialized preservation methods. Several representative strains with unique preparations are listed in Table 3; however, these examples and methods may not be applicable to all members of the group. Overall, the optimum formulations for individual viral strains must be determined empirically.
Table 3: Examples of frozen preparations
| ATCC number | Product name | Preparation |
|---|---|---|
| VR-1 | Human adenovirus 1 | Infected culture medium |
| VR-343 | Japanese encephalitis virus | Infected culture medium diluted 1:1 with calf serum |
| VR-977 | Human herpesvirus 5 | Infected culture medium (EMEM + 10% FBS + 7% DMSO) |
| VR-897 | Influenza A virus (H1N1) | Infected allantoic medium |
| VR-955 | Human respiratory syncytial virus | Infected culture medium (EMEM + 2% FBS) |
| VR-129B | Encephalomyocarditis virus | Infected culture medium (MEM + 2% FBS) |
| VR-838 | Raccoonpox virus | Infected culture medium (EMEM + 2% FBS) |
| VR-156 | Vaccinia virus | Infected culture medium (L-15 + 2% FBS + L-glut) |
| VR-659 | Rous sarcoma virus | Infected culture medium (M199) |
| VR-137 | Rabies street virus | 10% infected mouse brain suspension in PBS + 10% horse serum |
| VR-612 | Rickettsia akari | Infected yolk sac diluted 1:1 in sucrose-phosphate glutamate |
Equipment
- Cryopreservation vials
There are two materials to choose from for cryopreservation vials: glass or plastic. Glass vials are more difficult to work with: they need to be sterilized before use, they need to be sealed with a hot flame, and they can be difficult to open. However, they are considered fail-safe once properly sealed.
If cryopreservation in glass ampoules is not possible, plastic vials can be used. Plastic vials come in two varieties: those with an internal thread and silicone gasket, and those with an external thread. Vials with an internal-thread were the first commercially available, but have some disadvantages over the external-thread version. For example, while the silicone gasket provides an excellent seal, it needs to be tightened just right; the vial will leak if the seal is too tight or too loose. - Controlled-rate freezing chambers
For cell-associated viruses, there are several means to achieve a cooling rate of -1°C per minute. The best method involves the use of a computer controlled, programmable electronic freezing unit (such as Thermo Scientific CryoMed Freezers), which rigorously maintains this rate of cooling. This is the method used exclusively at ATCC. Such equipment is relatively expensive and necessary for only the most sensitive strains.
A less costly approach is to place the cryopreservation vials into an insulated chamber and cool for 24 hours in a mechanical freezer at -70°C or colder. There are several commercially available freezing chambers, which achieve a cooling rate very close to the ideal -1°C per minute (CoolCell LX; ATCC ACS-6000). Alternatively, the vials can be placed into a polystyrene box, with 15 mm (3/4 inch) thick walls and 1 L capacity that is packed with paper, cotton wool, or foam peanuts for insulation.
Liquid nitrogen freezing storage
The ultra-low temperatures (below -120°C) required for long-term storage can be maintained by specialized electric freezers, or more commonly, by liquid nitrogen freezers. There are two basic types of liquid nitrogen storage systems: immersing vials in the liquid or holding vials in the vapor phase above the liquid. The liquid-phase system holds more nitrogen and thus requires less maintenance. However, there is always a chance that some liquid will enter improperly sealed vials, which may then explode when retrieved. For this reason, ATCC strongly recommends storage in the vapor-phase.
Vapor-phase storage systems create a vertical temperature gradient within the container. The temperature in the liquid nitrogen at the bottom will be -196°C, whereas the temperature at the top will vary depending upon the amount of liquid nitrogen at the bottom and the length of time the container is opened. To ensure the safe storage of cells, maintain sufficient levels of liquid nitrogen in the container so that the temperature at the top is -120°C or colder. All storage systems should be equipped with temperature alarms.
Cryopreservation procedure
- Uncontrolled freezing
The viability of most viruses is best maintained by fast freezing after harvesting. Dispense the material into ampoules, place the ampoules in a rack, and dip in a dry ice/ethanol bath. Store the frozen material in liquid nitrogen vapor or in a mechanical freezer at -80°C. - Controlled freezing
To preserve cell-associated pathogens, such as Chlamydia and Rickettsia, a controlled freeze is preferred.- Incubate the viral agent under the recommended conditions until a maximum yield is obtained (maximum cytopathic effect if applicable).
- Add a cryoprotective agent to the viral suspension. For many viral strains, this will be a mixture of FBS and DMSO or glycerol. Some strains may require specialized conditions, examples of these can be found in Table 1.
- Within a biological safety cabinet, dispense 0.5 to 1.0 mL of the above mixture into sterile plastic cryovials or glass ampoules. Store the filled glass ampoules on ice prior to sealing. Glass ampoules can be sealed using a gas-oxygen torch, pulling the neck of the ampoules as it is rotated in the flame. Following sealing, disinfect glass ampoules by spraying with disinfectant.
- Place the vials into a pre-cooled (4°C), controlled-rate freeze chamber and place the chamber in a mechanical freezer at -70°C (or colder) for at least 24 hours. Alternately, use a pre-cooled (4°C) programmable freezer unit set to cool the vials at -1°C per minute until a temperature below -40°C is achieved and then set the temperature to abruptly drop to -120°C.
- Quickly transfer the vials to a liquid nitrogen or -120°C freezer. Frozen material will warm up above -50°C.
- Record the location and details of the freeze.
- After 24 hours at -120°C, remove one vial, appropriately restore the viral strain, and determine the viability and sterility.
Recovery of cryopreserved viruses
- In advance, grow the recommended production host in a prepared culture vessel that contains growth medium equilibrated for both temperature and pH.
- Remove the vial of frozen virus from the liquid nitrogen freezer and thaw by gentle agitation in a 37°C water bath.
- Thaw the strain rapidly until all ice crystals have melted (approximately 2 minutes).
- Remove the vial from the water bath and decontaminate it by dipping in or spraying with 70% ethanol. Follow strict aseptic conditions in a biological safety cabinet for all further manipulations.
- Unscrew the top of the vial and inoculate the propagation host with the recommended infection conditions as described on the supplied product sheet.
- Examine the cultures after an appropriate length of time.
Lyophilization
Overview
Freeze-drying is a process where water and other solvents are removed from a frozen product via sublimation.15 Sublimation occurs when a frozen liquid goes directly to a gaseous state without entering a liquid phase. The freeze-drying process results in a stable, readily rehydrated product. This process consists of three steps: pre-freezing the product to form a frozen structure, primary drying to remove most water, and secondary drying to remove bound water.
During the initial freezing process, ice crystals begin to form and the concentration of solutes in the suspension increases. The method used during freezing can greatly affect the ability to freeze-dry the material. Slow cooling rates are recommended as this will result in the formation of vertical ice crystal structures and allow for more efficient water sublimation from the frozen product.
Freeze-dried products are hygroscopic and must be protected from moisture during storage. Additionally, these products are sensitive to other factors including oxygen and temperature, which can significantly decrease the shelf life. It is important to store freeze-dried material in a manner that protects the product from exposure to moisture and oxygen and at refrigerated temperatures (4°C).
Generally, the lyophilization of viruses is not currently used by ATCC. This is because the methods for lyophilization vary depending on the type of virus, and some viruses cannot be successfully preserved by freeze-drying due to loss of viability. Strongly cell-associated viruses, including certain members of the herpes virus family, such as varicella-zoster, lose viability outside the host. Therefore, these viruses cannot be preserved by this method. Though ATCC does not currently freeze-dry any virus preparations, it was done fairly extensively in the past. The methods of lyophilization that were previously used by ATCC are described below. For additional information, contact ATCC technical services.
Equipment
- Lyophilization vials
For the storage of lyophilized viruses, ATCC uses sterile funnel-tipped glass ampoules (Wheaton) and glass serum vials. Generally, during the lyophilization process, material is freeze-dried in a glass ampoule, disinfected, and sealed. - Lyophilization apparatuses
For the lyophilization of viruses, ATCC previously employed a commercial freeze-dryer. In this freeze-drying procedure, samples are mixed with a suitable preservative, dispensed into the appropriate ampoule, and allowed to slowly freeze into a solid mass. The preservative used for lyophilization can vary between viral agents; several examples are listed below in Table 4. Once frozen, samples are lyophilized within a freeze-drying system (Virtis Genesis, Millrock Max 85, and LD 85).
During the primary drying phase, water is removed from the frozen product via sublimation. This is accomplished through the use of a vacuum pump, which allows water molecules to migrate from the frozen product and condense on a moisture trap called a condenser. For this to be possible the temperature of the condenser must be colder than the product temperature; the difference in these temperatures will affect the rate of sublimation. When primary drying is complete, all residual moisture is removed by directly heating the product. During this secondary drying phase, water must be desorbed to a residual moisture content of 1% or less. This process requires a low pressure, low condenser temperature system. Once dried, the ampoules are properly sealed and stored at refrigerated temperatures (4°C).
Table 4: Examples of freeze-dried preparations
| ATCC number | Product name | Preparation |
|---|---|---|
| VR-343 | Japanese encephalitis virus | Infected primary hamster kidney cells + 50% calf serum |
| VR-90 | Colorado tick fever virus | Infected mouse brain (10% suspension in normal saline) diluted 1:1 in rabbit serum |
| VR-897 | Influenza A virus | Infected allantoic fluid + 10% sucrose |
Storage and viability of lyophilized strains
To maximize the recovery of viable cells, viral cultures must be in optimum condition before the lyophilization process.16 Viruses should be propagated under the recommended conditions for infection as indicated on the product sheet.
Because lyophilized products are hygroscopic, they must be stored under moisture-free conditions. Additionally, other factors such as oxygen content and temperature can affect the shelf-life of freeze-dried strains. Oxygen can chemically react with the product, negatively affecting culture viability. This reactivity is directly proportional to storage temperature. Therefore, lyophilized products should be stored long-term in liquid nitrogen.
Lyophilization procedure

- Grow the virus according to usual procedures.
- For freeze-drying viral suspensions, glucose, skim milk, or Sucrose-Phosphate-Glutamate-Albumin (SPGA) should be added before dispensing. The lyophilization of some viruses may require specific preservation preparations; examples are listed in Table 4.
- In a biological safety cabinet, dispense the suspension into either sterile funnel-tipped ampoules or serum vials. Plug the ampoules loosely with slotted silicon rubber stoppers, and place the ampoules in a freeze-dryer tray. Set an acrylic plate on top of the stoppers.
- Freeze-dry the material in a commercial freeze-dryer. Hold the material at -30°C for 18-48 hours, depending on the additive. Then raise the shelf temperature to 10°C per hour until the product temperature is 25°C.
- When the cycles are complete, backfill the system with sterile nitrogen and press the caps into place. Disinfect the outside of the ampoules and return them to the biological safety cabinet.
- Seal the vials. Funnel-tipped ampoules will need to be sealed by an oxygen-gas torch.
Recovery of lyophilized cells
- To rehydrate freeze-dried strains, add sterile double-distilled water.
- Mix well, dilute with culture medium, and add the virus to the propagation host cell culture.
- Incubate strains under the recommended temperature and atmospheric conditions.
Preservation of specific strains
All viral strains can be cryopreserved and maintained in liquid nitrogen vapor, whereas only some strains can by lyophilized. In preparation for cryopreservation or lyophilization, viral strains should be grown under optimal conditions. This can include growth in chicken eggs or within tissue culture. Below, we provide information on the preservation of common viral strains.
Adenoviruses
Image of Adenovirus courtesy of Dr. G. William Gary, Jr., CDC
The less labile virus suspensions, such as human adenovirus, are harvested in their culture media then dispensed into ampoules for freezing.
Herpesviruses
Certain members of the herpes family, such as cytomegalovirus or varicella-zoster, lose viability outside the host. Therefore, keep infected cells intact prior to preservation.
Viruses grown in chicken embryos
Viruses harvested from chicken eggs as allantoic fluid or yolk sac preparations can be frozen or lyophilized. The high-protein content of the egg serves as a cryoprotectant, and other additives may not be necessary.
Viruses grown in vivo
Viruses grown in vivo and harvested in organ suspensions are preserved by freezing. Add sterile 50% glycerol or 10-50% serum as a cryoprotectant.
Special hazards
Care must be taken during the cryopreservation or lyophilization of animal viruses. Problems such as contamination, breakage of glass ampoules during handling and storage, dispersal of freeze-dried virus when opening glass ampoules, and the handling of liquid nitrogen must all be considered. To prevent contamination and the dispersal of viruses, aseptic technique must be followed. This can include the decontamination of all equipment and vials as well as performing all preparations in a biological safety cabinet. Additionally, protective clothing should be worn during preparation to prevent contamination as well as to guard against harm due to contact with liquid nitrogen.

Biosafety and disposal
Biosafety
The need for precautions when experimenting with viral cultures depends upon the source and nature of the biological material, the experimental procedure, and the laboratory/containment conditions. Since every situation is different, the risks need to be identified for each individual strain and the appropriate precautions need to be taken before any work begins.
More information on risk assessment and precautions can be found in the Center for Disease Control (CDC) publication Biosafety in Microbiological and Biomedical Laboratories.17 The text of this publication is available in its entirety online at www.cdc.gov.
A biosafety level (BSL) is assigned to each viral strain for the purposes of packaging for safe shipment. ATCC follows federal biosafety guidelines and takes several factors into consideration when assessing a potential hazard, and in some cases the ATCC assigned biosafety level is more restrictive. Generally, ATCC only ships and stores viruses with a biosafety level assignment of 1, 2, or 3.
Biosafety level 1
- Work involving well-characterized viral strains not known to consistently cause disease in immunocompetent adult humans.
- Work can be conducted on the bench top using aseptic technique, no special containment equipment or facility is required.
Biosafety level 2
- Work involving viral strains that pose a moderate hazard to health adult humans.
- Work should be conducted in designated biological safety cabinets within laboratories with restricted access.
Biosafety level 3
- Work involving indigenous or exotic agents that may cause serious or potentially lethal disease via inhalation.
- Work should be conducted in biological safety cabinets localized inside a specialized BSL-3 containment facility within laboratories with restricted access.
As the recipient of an ATCC virus, take into account not only the nature of the material but also the manipulations employed during its handling when assessing the potential laboratory risk. Keep in mind that there will be situations where the intended use of an agent may require more stringent precautions than associated with the assigned biosafety level.17
Disposal of infectious materials
All viral cultures, stocks, and potentially infectious materials need to be properly decontaminated prior to disposal. The written method for proper decontamination should be available in the laboratory and BSL facility. Several methods of sterilization include use of an autoclave, chemical disinfection, incineration, or any other validated decontamination method. More information on the disposal of viral cultures can be found in the Center for Disease Control (CDC) publication Biosafety in Microbiological and Biomedical Laboratories.17
Viral authentication and viability testing
When preparing a viral strain for distribution, ATCC performs numerous quality control (QC) assays to guarantee that the product is of the highest standard before it reaches the consumer. All viral strains grown in-house undergo viability testing as well as genotypic examinations to ensure that the strain identification is accurate and the viral agent is sustainable. Described below are several tests used by ATCC for the authentication and viability testing of animal viruses.
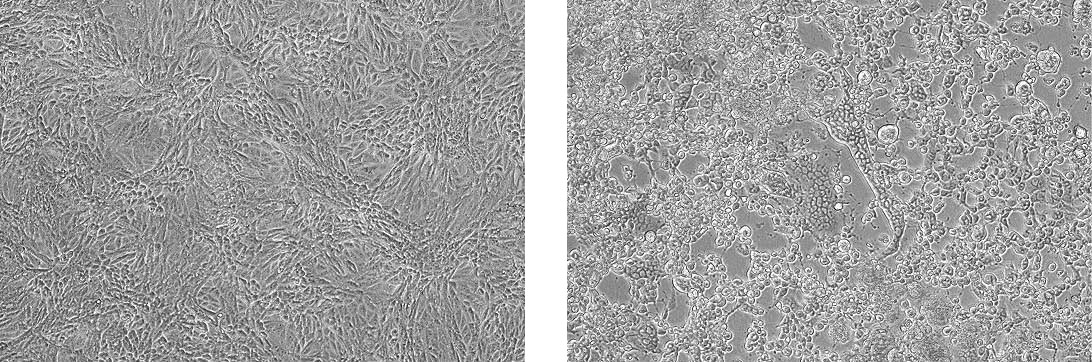
Propagation of Human herpes virus 1 (ATCC VR-260) in Vero cells (ATCC CCL-81). The left panel depicts uninfected CCL-81 cells. The right panel depicts infected CCL-81 cells exhibiting CPE.
Viability testing
Cytopathic effects (CPE)
For tissue-culture adapted viruses, viability is primarily monitored by observing host cell appearance. During the synthesis of viral components, the host cell undergoes morphological and biochemical changes. These deviations often result in degenerative changes known as cytopathic effect. The degree of visible cell damage varies between viral species and host cell lines. Some viruses cause little to no CPE in host cells, while others can destroy the host cell population. Characteristic CPE is best observed by analyzing cultures daily.18 (See NOTE 6)
NOTE 6:
During early viral infection, cells may become rounded or appear more refractive. More severe CPE include focal degeneration, vacuolization, cellular fusion, or host cell death.
Hemagglutination
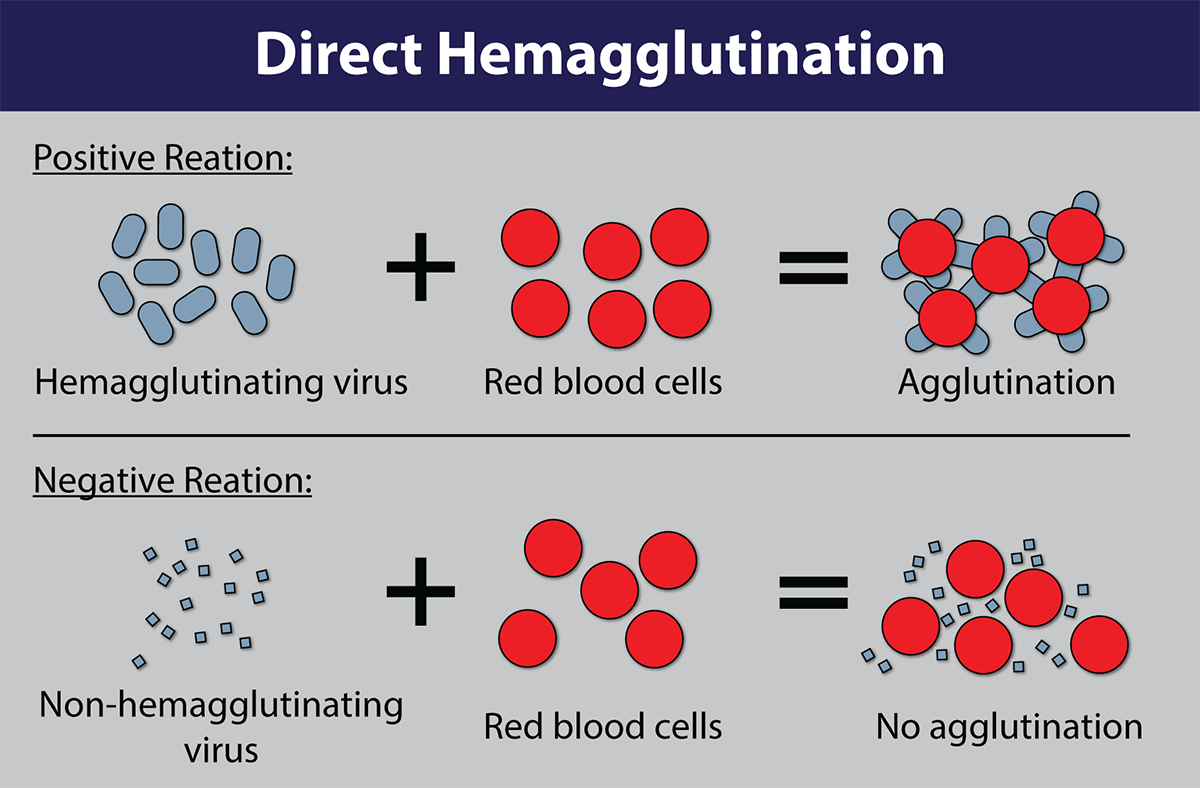
Image of Hemagglutination assay courtesy of Dr. F.T. Forrester., CDC
Many viruses can attach to molecules present on the surface of red blood cells. At certain concentrations, a viral suspension may cause red blood cells to clump together. This natural phenomenon, known as hemagglutination, prevents red blood cells from settling out of suspension and has been adapted for use in viability testing and CEID viral titering. In a hemagglutination assay, chicken red blood cell suspensions are incubated with a serially diluted virus and monitored for the formation of a red blood cell lattice. The end-point is taken to be the highest dilution of the virus suspension that produces a positive result.
Hemadsorption assay
The phenomenon of hemadsorption is dependent on the attachment of red blood cells to the surface of cell monolayers infected with enveloped, hemagglutinin-producing viruses. This natural process can be adapted as a general procedure to determine viral potency. In a hemadsorption assay, a red blood cell suspension is incubated with an infected cell culture. If the cell monolayer is infected with a hemagglutinin-producing virus, hemagglutinin is inserted into the cell plasma membrane during viral reproduction in preparation for viral maturation. It is at these modified areas of the cell surface that red blood cells will specifically bind. Thus, hemadsorption is indicative of the presence of viruses that produce hemagglutinin, such as influenza, measles, mumps, and parainfluenza.
Viral infectivity
Viral infectivity is often measured by calculating titer. Depending on the required host for viral propagation, infectivity can be analyzed using a plaque assay or by determining the infectious dose in tissue culture or embryonated chicken eggs. The basis of these assays is to measure the ability of an infectious viral suspension to produce a pathological change in the host environment. For additional information on plaque assays or calculating infectious dose, refer to the chapter Viral Replication and Propagation.
Viral authentication
Sequencing
The ATCC Virology collection utilizes sequencing to authenticate our holdings as they are accessioned or come up for replenishment. ATCC sequences approximately ≥500 bp of the viral genome that is characteristic of the species or virus type.
Enzyme linked immunosorbant assay
All viruses have at least one unique surface antigen. Through an enzyme-linked immunosorbant assay (ELISA), these antigens can be used as a biological marker to authenticate a viral species. There are two forms of this assay, the direct and indirect ELISA. Both assays employ enzyme-conjugated monoclonal detection antibodies that tightly bind a specific viral antigen. With a direct ELISA, one is testing for the presence of the virus, while an indirect ELISA assay detects specific viral antibody rather than the virus itself. The presence of a specific virus is analyzed by assessing the conjugated enzyme activity via incubation with a substrate that is converted into a measurable colored product. When using an ELISA for authentication purposes, a colorimetric change indicates the viral sample was correctly identified.
Viral applications
Cell transformation
Cancerous cells emerge due to discrete changes in the cellular genome. These genomic deviations can arise due to viral transformation. During the infection of non-permissive cells, viral genetic material can become covalently integrated into the host genome. If viral integration induces the aberrant expression of a proto-oncogene, cell growth can become unrestricted, resulting in an immortal cell state.
This natural process has been adapted by cell biologists for use in the immortalization of mammalian cells in tissue culture. Immortalized cell lines offer the possibility of an inexhaustible supply of cells, allowing for more consistent and reproducible research. Viral genes such as the simian virus 40 (SV40) T antigen have been used as a simple, reliable agent for the immortalization of a variety of cell types.19,20 SV40 is believed to induce immortalization by suppressing the transcription of p53, a tumor suppressor protein responsible for initiating apoptosis following cell damage.
In addition to their use in cell immortalization, viral strains can be used to introduce nucleic acids into cells. This process, termed transduction, allows scientists to analyze the function of specific genes, to examine the effects of gene silencing, or to introduce therapeutic genes.
Phage therapy
Bacteriophages are viruses that infect bacterial cells, often resulting in the lysis and subsequent death of the bacterial host. Since the early 20th century, European scientists have taken advantage of this natural process to treat bacterial infections in a process known as phage therapy. Because the host range of a bacteriophage is very narrow, often specific to one species, they are considered more effective than antibiotic treatment and result in less harm to the human microflora.22 Though phage therapy has many benefits, it was abandoned by many countries due to the antigenic properties and unpredictable behavior of bacteriophages in addition to the wide-spread production and use of antibiotics. However, with the recent emergence of antibiotic-resistant bacteria, scientists are now forced to re-evaluate the beneficial properties of the application of phage therapy.23
Nanotechnology
Cancer treatments have commonly employed surgery, radiation, chemotherapy, and hormone therapy.24 Though these treatments have helped millions of patients go into remission, they come at a cost. Most cancer treatments attack both healthy and cancerous cells, often leaving patients nauseous, fatigued, and immunocompromised. To combat these negative side effects, viral nanoparticles are being engineered and analyzed as a potential form of drug delivery.
Viral nanoparticles are viruses whose genomic material has been removed and replaced with therapeutic drugs. Because viruses have naturally evolved the ability to cross the host cell membrane, they are an ideal candidate for drug packaging and targeted delivery. To minimize toxic side effects, infection, and induced immune response, human viruses are not used in the production of viral nanoparticles. Rather, these drug delivery systems are engineered from plant, insect, and animal viruses.25 Plant viruses, such as the Cowpea mosaic virus, are ideal candidates for nanotechnology as they are easy to produce in large quantities, can self-assemble around a nanoparticle, and hold a substantial volume of cancer drugs.26
One of the major benefits to using viral nanoparticles for drug delivery is that targeting molecules can be easily attached to the viral surface, allowing for the directed binding to cancer cells. This specific binding prevents the harm or death of surrounding healthy cells, thus reducing possible side effects. Though viral nanoparticles have the potential to be a beneficial treatment, there are several complications associated with their use as a drug delivery system including immune rejection and potential toxicity. Regardless, viral nanoparticles have the capability to revolutionize the treatment of cancer, creating a safe and specific form of drug delivery.
Recombinant vectored vaccines
Vaccines are the most effective prophylactic tool for the prevention of disease. Viral-based vaccines are traditionally employed as live attenuated viruses or as chemically inactivated viruses. Though these types of preparations have been shown to induce protective immunity in animal models, incomplete viral attenuation or inactivation poses a serious risk to those who are vaccinated.27,28 Recombinant vectored vaccines, however, offer a live-vaccine approach that does not employ the complete pathogen. Instead, genetic material from the microbial target strain is inserted into the genome of harmless viral strains, allowing for the safe expression of microbial antigens.
Newcastle disease virus (NDV), a negative-sense RNA avian virus, is a common template used for the development of recombinant vectored vaccines. Not only does NDV grow to high titers in many cell lines and eggs, it can elicit a strong immune response in vivo, is harmless to humans, and is commercially available. Additionally, NDV replicates in the cytoplasm of infected cells, thus eliminating the problem of genetic integration.29 To create a viral vaccine vector from NDV, genes encoding foreign target proteins are inserted into the NDV genome through recombination. Upon vaccination with the live virus, the foreign target protein is expressed within the host cell cytoplasm where it is then available for processing by the cellular antigen-processing machinery for immune presentation. As a result, cellular immunity is activated and neutralizing antibodies are generated.30-32
Get a copy of the ATCC Virology Culture Guide to help with your research
Download the Guide

Appendix
Table 5: Propagation hosts and recommended basal media
| ATCC number | Product description | ATCC number | Media |
|---|---|---|---|
| CCL-22 | MDBK (NBL-1) | 30-2003 | Eagle’s Minimum Essential Medium |
| CCL-34 | MDCK (NBL-2) | 30-2003 | Eagle’s Minimum Essential Medium |
| CRL-1590 | SL-29 | 30-2002 | Dulbecco’s Modified Eagle’s Medium |
| CCL-10 | BHK-21 | 30-2003 | Eagle’s Minimum Essential Medium |
| CCL-61 | CHO-K1 | 30-2004 | F-12K Medium |
| CRL-1573 | 293 (HEK-293) | 30-2003 | Eagle’s Minimum Essential Medium |
| CCL-2 | HeLa | 30-2003 | Eagle’s Minimum Essential Medium |
| CCL-23 | Hep-2 | 30-2003 | Eagle’s Minimum Essential Medium |
| CCL-171 | MRC-5 | 30-2003 | Eagle’s Minimum Essential Medium |
| CCL-75 | WI-38 | 30-2003 | Eagle’s Minimum Essential Medium |
| CRL-1721 | Sf9 | 30-2001 | RPMI-1640 Medium |
| CRL-1651 | COS-7 | 30-2002 | Dulbecco’s Modified Eagle’s Medium |
| CCL-81 | Vero | 30-2003 | Eagle’s Minimum Essential Medium |
| CRL-1586 | VERO C1008 [Vero 76, clone E6, Vero E6] | 30-2003 | Eagle’s Minimum Essential Medium |
| CRL-1587 | VERO 76 | 30-2002 | Dulbecco’s Modified Eagle’s Medium |
| CCL-7.1 | LLC-MK2 Derivative | 30-2003 | Eagle’s Minimum Essential Medium |
| CL-160 | DBS-FRhL-2 | 30-2003 | Eagle’s Minimum Essential Medium |
| CCL-33 | PK(15) | 30-2003 | Eagle’s Minimum Essential Medium |
ATCC Reagents for Virus Expansion
Table 6: ATCC cell lines for virus expansion
| ATCC number | Product name | Organism | Tissue source |
|---|---|---|---|
| CCL-22 | MDBK | Bovine | Kidney |
| CCL-34 | MDCK (NBL-2) | Canine | Kidney |
| CRL-1590 | SL-29 | Chicken | Embryo |
| CCL-10 | BHK-21 | Hamster | Kidney |
| CCL-61 | CHO-K1 | Hamster | Ovary |
| CRL-1573 | 293 (HEK-293) | Human | Kidney |
| CRL-1573.3 | HEK-293.2sus | Human | Kidney |
| CCL-2 | HeLa | Human | Cervix |
| CCL-23 | Hep-2 | Human | Cervix |
| CCL-171 | MRC-5 | Human | Lung |
| CCL-75 | WI-38 | Human | Lung |
| CRL-1721 | Sf9 | Insect | Ovary |
| CRL-1651 | COS-7 | Monkey | Kidney |
| CCL-81.5 | Vero-SF-ACF | Monkey | Kidney |
| CCL-81 | Vero | Monkey | Kidney |
| CRL-1586 | VERO C1008 [Vero 76, clone E6, Vero E6] | Monkey | Kidney |
| CRL-1587 | VERO 76 | Monkey | Kidney |
| CCL-7.1 | LLC-MK2 Derivative | Monkey | Kidney |
| CL-160 | DBS-FRhL-2 | Monkey | Lung |
| CCL-33 | PK(15) | Pig | Kidney |
Table 7: ATCC media product list
| ATCC number | Product name | Volume |
|---|---|---|
| 30-2002 | Dulbecco's Modified Eagle's Medium (DMEM) | 500 mL |
| 30-2006 | DMEM:F12 Medium (1:1 Ratio) | 500 mL |
| 30-2003 | Eagle's Minimum Essential Medium (EMEM) | 500 mL |
| 30-2004 | F12K Medium | 500 mL |
| 30-2005 | Iscove's Modified Dulbecco's Medium (IMDM) | 500 mL |
| 30-2008 | Leibovitz's L-15 Medium | 500 mL |
| 30-2007 | McCoy's 5A Medium, Modified | 500 mL |
| 30-2001 | RPMI-1640 Medium | 500 mL |
Table 8: Animal sera product list
| ATCC number | Product name | Volume |
|---|---|---|
| 30-2020 | Fetal Bovine Serum | 500 mL |
| 30-2030 | Calf Bovine Serum | 500 mL |
| 30-2040 | Horse Serum | 500 mL |
Table 9: Cell culture supplements
| ATCC number | Product name |
|---|---|
| Amino acid solutions | |
| 30-2214 | L-Glutamine Solution, 200 mM |
| Subculture reagents | |
| 30-2101 | Trypsin EDTA Solution |
| 30-2103 | Non-Enzymatic Cell Dissociation Solution |
| 30-2104 | Soybean Trypsin Inhibitor |
| Buffers, stains & water | |
| 30-2200 | Dulbecco's Phosphate Buffered Saline (DPBS) |
| Cryopreservation reagents and tools | |
| 4-X | Dimethylsulfoxide (DMSO) |
| 30-2600 | Serum-Free Cell Freezing Medium |
| ACS-6000 | CoolCell LX Alcohol-free Freezing Container |
Table 10: Cell proliferation assays and mycoplasma detection
| ATCC number | Product name |
|---|---|
| 30-1010K | ATCC MTT Cell Proliferation Assay |
| 30-1011K | XTT Cell Proliferation Assay Kit |
| 30-1012K | Universal Mycoplasma Detection Kit |
Glossary
Ambisense. A term applied to single-stranded RNA viral genomes. Part of the nucleotide sequence is positive-sense and part is negative-sense.
Complex morphology. A virus that possesses a capsid that is neither truly helical nor icosahedral. This morphology may have additional structures such as a protein tail or a complex outer wall.
Cytopathic effect (CPE). CPE refers to the degenerative changes in association with viral infection.
Enveloped morphology. Viral envelopes are commonly derived from portions of the host cell membranes that have incorporated viral glycoproteins. Envelopes surround the viral capsid and assist in viral entry of host cells.
Helical morphology. Viral morphology composed of a single type of capsomer stacked around a central axis to form a helical structure.
Hemagglutination. This term refers to the agglutination, or clumping, of red blood cells.
Icosahedral morphology. Viral structures built from repeated identical protein subunits to form a nearly spherical structure with rotational symmetry.
Multiplicity of infection (MOI). The ratio of agents (eg, viruses, bacteriophages, bacteria) to infection targets (eg, propagation host).
Negative sense. Negative-sense RNA (3’ to 5’) forms the complementary strand and must be converted to positive-sense by an RNA polymerase prior to translation.
Non-permissive. Being or relating to a cell or environmental condition that does not support the replication of a virus or bacteriophage.
Positive sense. Positive-sense RNA (5’ to 3’) signifies that a particular sequence can be directly translated into protein.
Production host. The host strain used for the propagation of viruses.
Prolate morphology. Viral morphology composed of a cylinder with a cap at either end forming an elongated icosahedron structure along a fivefold axis. This is a common arrangement of bacteriophage heads.
Proto-oncogene. This term defines a normal gene that can become an oncogene due to mutations or increased expression. These genes commonly code for proteins involved in the regulation of cell growth or differentiation.
Sense. Sense is a concept used to compare the polarity of nucleic acids.
Transduction. The process by which genetic material is introduced into a cell by a virus or viral vector.
Transformation. This term refers to the cellular alteration resulting from the uptake, integration, and expression of exogenous genetic material taken up from the environment.
References
- Rovozzo, G. & Burke, C. A Manual of Basic Virological Techniques (Prentice-Hall, 1973).
- Leland, D. & Ginocchino, C. Role of Cell Culture for Virus Detection in the Age of Technology. Clin Microbiol Rev 20, 49-78 (2007).
- Cohen, J.I. Epstein-Barr virus infection. N Engl J Med 343, 481-92 (2000).
- Gotoh, K. et al. Replication of Epstein-Barr virus primary infection in human tonsil tissue explants. PLoS One 6, e25490.
- Mahy, B. & Kangro, H. Virology Methods manual (Academic Press, 1996).
- Reed, L. & Muench, H. A simple method of estimating fifty percent endpoints. American Journal of Hygiene 27, 493-497 (1938).
- McLimans, W. (eds. Rothblat, G. & Cristofalo, V.) (Academic Press, New York, 1972).
- Shipman, C., Jr. Evaluation of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) as a tissue culture buffer. Proc Soc Exp Biol Med 130, 305-10 (1969).
- Giandomenico, A.R., Cerniglia, G.E., Biaglow, J.E., Stevens, C.W. & Koch, C.J. The importance of sodium pyruvate in assessing damage produced by hydrogen peroxide. Free Radic Biol Med 23, 426-34 (1997).
- Jacoby, W. & Pasten, I. (Academic Press, New York, 1979).
- Mazur, P. The role of intracellular freezing in the death of cells cooled at supraoptimal rates. Cryobiology 14, 251-72 (1977).
- Mazur, P. Cryobiology: the freezing of biological systems. Science 168, 939-49 (1970).
- Fahy, G.M. The relevance of cryoprotectant "toxicity" to cryobiology. Cryobiology 23, 1-13 (1986).
- Meryman, H.T. Cryoprotective agents. Cryobiology 8, 173-83 (1971).
- Rowe, T.W. Optimization in freeze-drying. Dev Biol Stand 36, 79-97 (1976).
- Heckly, R. Principles of preserving bacteria by freeze-drying. Dev Ind Microbiol 26, 379-395 (1985).
- United States Department of Health and Human Serves, C.f.D.C., and National Institutes of Health. Biosafety in Microbiological and Biomedical Laboratories (U.S. Government Printing Office, HHS Publication No. (CDC) 21-1112, Washington DC, 2009).
- Enders, J.F. Cytopathology of virus infections: particular reference to tissue culture studies. Annu Rev Microbiol 8, 473-502 (1954).
- Jha, K.K., Banga, S., Palejwala, V. & Ozer, H.L. SV40-Mediated immortalization. Exp Cell Res 245, 1-7 (1998).
- Kirchhoff, C. et al. Immortalization by large T-antigen of the adult epididymal duct epithelium. Mol Cell Endocrinol 216, 83-94 (2004).
- Mancheno-Corvo, P. & Martin-Duque, P. Viral gene therapy. Clin Transl Oncol 8, 858-67 (2006).
- Miedzybrodzki, R. et al. Clinical aspects of phage therapy. Adv Virus Res 83, 73-121.
- Kropinski, A.M. Phage Therapy - Everything Old is New Again. Can J Infect Dis Med Microbiol 17, 297-306 (2006).
- Institute, N.C. (2011).
- Singh, P., Destito, G., Schneemann, A. & Manchester, M. Canine parvovirus-like particles, a novel nanomaterial for tumor targeting. J Nanobiotechnology 4, 2 (2006).
- Franzen, S. & Lommel, S.A. Targeting cancer with 'smart bombs': equipping plant virus nanoparticles for a 'seek and destroy' mission. Nanomedicine (Lond) 4, 575-88 (2009).
- Brown, F. Review of accidents caused by incomplete inactivation of viruses. Dev Biol Stand 81, 103-7 (1993).
- Nathanson, N. & Langmuir, A.D. The Cutter Incident. Poliomyelitis Following Formaldehyde-Inactivated Poliovirus Vaccination in the United States during the Spring of 1955. Ii. Relationship of Poliomyelitis to Cutter Vaccine. Am J Hyg 78, 29-60 (1963).
- Huang, Z., Elankumaran, S., Panda, A. & Samal, S.K. Recombinant Newcastle disease virus as a vaccine vector. Poult Sci 82, 899-906 (2003).
- Huang, J.L. et al. High-level expression of recombinant dengue viral NS-1 protein and its potential use as a diagnostic antigen. J Med Virol 65, 553-60 (2001).
- Krishnamurthy, S., Huang, Z. & Samal, S.K. Recovery of a virulent strain of newcastle disease virus from cloned cDNA: expression of a foreign gene results in growth retardation and attenuation. Virology 278, 168-82 (2000).
- Nakaya, T. et al. Recombinant Newcastle disease virus as a vaccine vector. J Virol 75, 11868-73 (2001).